
Omics-Analyse
(Immunantwort auf Infektionen)
(Stand: Januar 2016)
Zur Beantwortung biologischer Fragestellungen werden häufig Experimente durchgeführt, bei denen unter Variation bestimmter Bedingungen eine Vielzahl von Daten erhoben werden. Handelt es sich um eine umfangreiche Aufnahme von Daten "einer Klasse", wird dann von "-omics" gesprochen, (z. B. Transcriptomics oder Secretomics). Nach der Erhebung müssen diese Daten analysiert werden, wofür BioControl über den biologisch-technischen Hintergrund, die geeigneten Algorithmen und gut interpretierbare Ergebnisdarstellungen verfügt. Im Folgenden wird dies am Beispiel einer Transkriptom-Analyse und ihrer Fragestellung zur Immunantwort auf Infektionen gezeigt.
Wichtig: bei ähnlichen Analysen werden durch BioControl die Methoden und Analysen in Absprache mit den experimentellen Partnern an das jeweilige Problem angepasst.
Fragestellung
Die biologischen Fragestellungen hinter einer Omics-Analyse betreffen ausgewählte Prozesse und ihre zugrundeliegenden Mechanismen. Im Allgemeinen geht es um die Reaktion biochemischer Komponenten auf unterschiedliche Bedingungen (z. B. Mutation, Umweltbedingungen, Erreger).
Das vorliegenden Beispiel analysiert reale Transkriptom-Daten, die mit Illumina Microarrays ("Illumina HumanHT-12 V4.0 expression beadchip") gewonnen wurden und öffentlich nutzbar sind:
[1] Sanne P. Smeekens et al.: "Functional genomics identifies type I interferon pathway as central for host defense against Candida albicans." In: Nat Commun, 4:1342, 2013. doi: 10.1038/ncomms2343, GEO: GSE42606.
Es geht um die Antwort des durch mononukleäre Zellen des peripheren Blutes (PBMC) repräsentierten Immunsystems auf verschiedene Bedingungen, insbesondere mit dem Schwerpunkt auf die Infektion mit Candida albicans. Dabei steht u.a. der Pathway des Typ 1 Interferons im Mittelpunkt. In der Studie sind Replikate von nicht-infiziertem Blut (30+35) sowie Infektionen bzw. Stimulation mit Borrelien (31+36), Candida (24+34), LPS (26+24) und Tuberkulose (23+36) zu 4h und 24h enthalten. Es werden im Folgenden nicht die Ergebnisse der genannten Publikation dupliziert (insbesondere nicht der Vergleich zwischen gesunden und kranken Spendern), sondern eine Auswahl an eigenen Analyseergebnissen gezeigt.
Methoden und Ergebnisse
Im folgenden werden die einzelnen Analyseschritte vorgestellt. Sie umfassen die messmethoden-spezifische Datenvorverarbeitung, statistische Tests für die differentielle Veränderungen von Transkripten und ein "Enrichment", also die Bestimmung von signifikant auftretenden Kategorien. Auf die Vorstellung einer Netzwerkinferenz, siehe [2, 3], sowie weiterer (z. B. in der Original-Publikation gezeigter) Ergebnisse wird an dieser Stelle verzichtet.
Datenvorverarbeitung
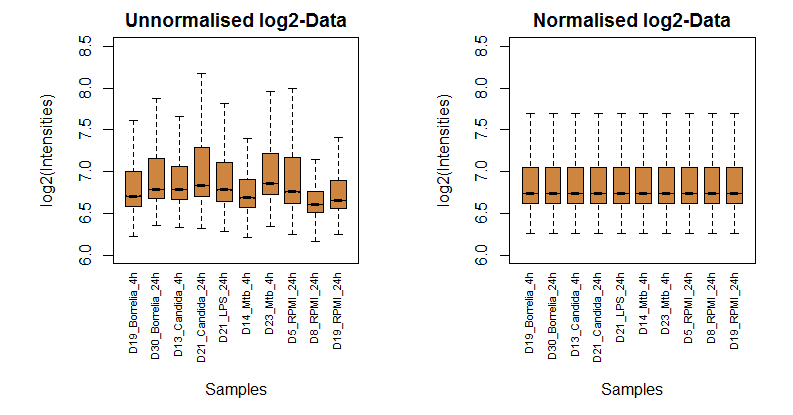
In diesem ersten Schritt werden messmethoden-spezifische Vorverarbeitungsschritte durchgeführt. Sie hängen von der Art des Experiments, der Messplattform (für Microarrys z. B. Affymetrix oder Illumina) und den damit verbundenen Daten- bzw. Fehlermodellen ab. Im vorliegenden Beispiel wird unter der Annahme, dass die Proben, also das Transkriptom der einzelnen Illumina Microarrays, eine ähnliche Verteilung (der Quantile) besitzen müssen, die entsprechende Normalisierung durchgeführt, siehe [4]. Erst nach diesem Normalisierungsschritt können die signifikant veränderten Komponenten bestimmt werden.
In den folgenden zwei Abbildungen werden die Verteilungen von 10 zufällig ausgewählten Proben vor und nach der Normalisierung miteinander verglichen:
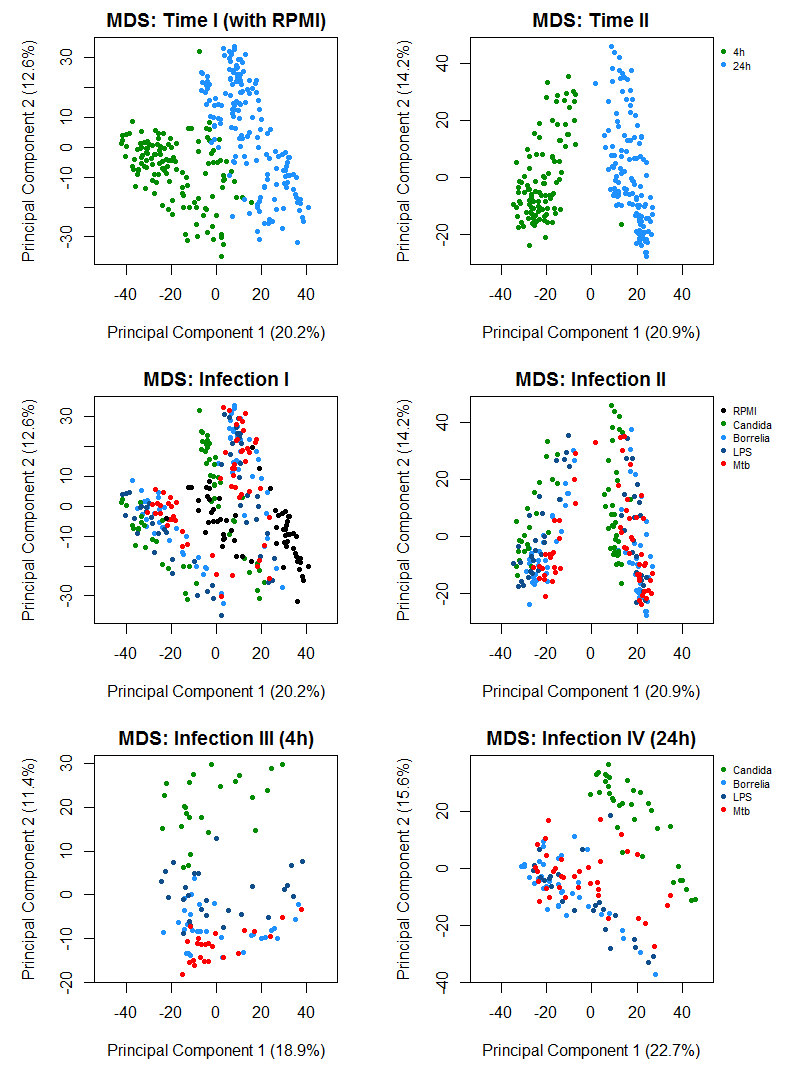
Für eine erste Auswertung der Ähnlichkeit zwischen den Proben bzw. definierten Gruppen können die ersten beiden Hauptkomponenten dieser normalisierten Daten dargestellt werden. Dafür wird hier die Methode "Multi-Dimensional Scaling" eingesetzt, siehe [5]. In den Abbildungen (gefärbt nach Zeitpunkt oder Infektion, mit und ohne Kontrolle) ist erkennbar, wie sich die Daten nach Mess-Zeitpunkt bzw. Infektion trennen lassen (wobei (1) die Trennung nach der Zeit am stärksten ist, (2) die Spender selbst einen großen Einfluss haben und (3) sich Candida am besten von den anderen 3 Bedingungen trennen lässt):
Statistische Tests
(Differentielle Expression und spezifische Antworten)
Mithilfe statistischer Tests können die Daten auf signifikant veränderte Komponenten (im Vergleich zur Kontrolle) untersucht werden. Die Ergebnisse bilden dann die Grundlage sowohl für eine Analyse dieser Komponenten (Namen, Anzahl, Intensität) als auch für weiterführende Untersuchungen ("Enrichment" beziehungsweise funktionelle Annotierung).
Den Ausgangspunkt der statistischen Tests stellen die normalisierten und logarithmierten Daten dar. Mithilfe der einzelnen Replikate und unter bestimmten Modellannahmen (hier: lineares Modell, normalverteilter Fehler) können die verschiedenen Bedingungen miteinander verglichen werden. Im vorliegenden Fall werden die Infektions-Bedingungen zu ihren entsprechenden Kontrollen der jeweiligen Zeitpunkte (4h und 24h) verglichen (da auch die Kontrolle sich mit der Zeit verändert, wie in den oben gezeigten Hauptkomponenten sichtbar ist)
Als Testmethode wird hier "TREAT" angewendet, siehe [6]. Es werden Schwellwerte für Foldchange (hier: 1.1) und adjustierten p-Wert (hier: 0.05, siehe [7]) benutzt. Diese Methode ist als eine Variante in dem R-Paket für lineare Modelle "limma" enthalten, siehe [8]. Als Ergebnisse sollen hier Heatmaps, Mengendarstellungen und Ergebnistabellen gezeigt werden.
Begonnen wird mit den Zahlenwerten für Gene, welche nach Infektion mit Candida am stärksten differentiell exprimiert sind:
| ProbeID | EntrezID | SymbolID | fc | padj |
|---|---|---|---|---|
| ILMN_2218856 | 414062 | CCL3L3 | 48.79 | 1.51E-55 |
| ILMN_2207291 | 3458 | IFNG | 44.71 | 1.27E-64 |
| ILMN_1728106 | 7124 | TNF | 44.45 | 1.16E-66 |
| ILMN_1747355 | 414062 | CCL3L3 | 42.47 | 1.41E-50 |
| ILMN_1671509 | 6348 | CCL3 | 41.25 | 1.52E-48 |
| ... | ... | ... | ... | ... |
| ILMN_2197365 | 5997 | RGS2 | -3.25 | 5.80E-25 |
| ILMN_1774761 | 729230 | CCR2 | -3.60 | 1.01E-14 |
| ILMN_1663866 | 7045 | TGFBI | -3.75 | 1.41E-07 |
| ILMN_1686623 | 1436 | CSF1R | -3.94 | 6.55E-12 |
| ILMN_1687301 | 1462 | VCAN | -4.57 | 6.01E-11 |
Nun sollen die Mengen bzw. die Anzahl von Genen bestimmt werden, welche für die verschiedenen Bedingungen SPEZIFISCH exprimiert sind. Dafür werden im Folgenden zwei unterschiedliche Ergebnisdarstellungen vorgestellt. Die erste Darstellungsform ist ein herkömmliches Venn-Diagramm basierend auf der Aussage, welche Gene im Vergleich zur Kontrolle verändert sind. Die zweite ist eine matrixartige Darstellung, die auch für viele Bedingungen eine gute Übersicht bietet, basierend auf der Aussage, welche Gene auch ZWISCHEN allen Bedingungen differentiell verändert sind.
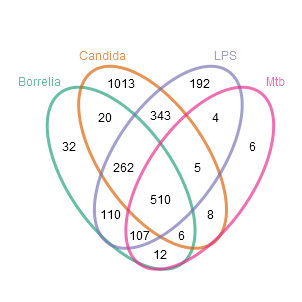
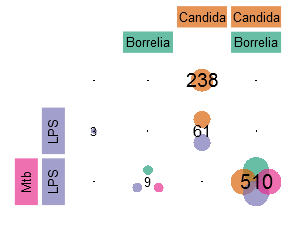
Hier ist zum einen erneut erkennbar, dass bei einer Infektion mit Candida im Vergleich zu den anderen Bedingungen viele spezifische Gene existieren, aber zum anderen auch sehr viele Gene (510) in allen Bedingungen gemeinsam differentiell exprimiert sind. Auf der Basis der Ergebnisse der zweiten Darstellung, können die Daten auch als Heatmap (hier herunterladen) mit Dendrogrammen zum Erkennen von Gengruppen gemeinsamer Eigenschaften dargestellt werden. Auch hier ist zu sehen, dass im Gegensatz zu den anderen Bedingung viele Gene nur bei Candida hochreguliert sind (prominente Beispiele: CCL8, CXCL10 und TNFSF13B), es aber noch mehr Gene - beispielsweise CCL3L3, TNF, CCL3 oder CCL3L1 - einer gemeinsamen Antwort gibt.
Neben den Abbildungen sind nun auch die eigentlichen Zahlenwerte interessant. Dafür werden im Folgenden die allgemeine und Candida-spezifische Antwort der 10 am stärksten (bezogen auf Candida, hoch- und runterexprimiert) differentiell exprimierten Gene gezeigt:
| ProbeID | EntrezID | SymbolID | fc_Borrelia_4h | fc_Candida_4h | fc_LPS_4h | fc_Mtb_4h | padj_Borrelia_4h | padj_Candida_4h | padj_LPS_4h | padj_Mtb_4h |
|---|---|---|---|---|---|---|---|---|---|---|
| ILMN_2218856 | 414062 | CCL3L3 | 18.91 | 48.79 | 36.07 | 15.65 | 1.57E-40 | 1.51E-55 | 5.80E-51 | 4.28E-32 |
| ILMN_1728106 | 7124 | TNF | 6.15 | 44.45 | 6.51 | 4.26 | 2.87E-24 | 1.16E-66 | 5.38E-24 | 9.02E-14 |
| ILMN_1747355 | 414062 | CCL3L3 | 20.75 | 42.47 | 35.94 | 18.07 | 2.30E-40 | 1.41E-50 | 1.86E-48 | 6.09E-33 |
| ILMN_1671509 | 6348 | CCL3 | 23.14 | 41.25 | 37.34 | 20.40 | 4.71E-41 | 1.52E-48 | 1.01E-47 | 5.97E-34 |
| ILMN_1773245 | 6349 | CCL3L1 | 12.05 | 40.45 | 28.13 | 10.95 | 7.12E-24 | 2.57E-41 | 3.59E-36 | 3.09E-19 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| ILMN_1769895 | 729230 | CCR2 | -2.90 | -3.25 | -3.05 | -2.48 | 1.85E-12 | 5.11E-14 | 7.84E-13 | 1.33E-07 |
| ILMN_2197365 | 5997 | RGS2 | -2.32 | -3.25 | -2.77 | -2.36 | 2.10E-14 | 5.80E-25 | 1.29E-19 | 9.48E-13 |
| ILMN_1774761 | 729230 | CCR2 | -3.06 | -3.60 | -3.27 | -2.55 | 2.67E-12 | 1.01E-14 | 6.68E-13 | 3.20E-07 |
| ILMN_1663866 | 7045 | TGFBI | -4.77 | -3.75 | -7.67 | -3.13 | 2.34E-11 | 1.41E-07 | 1.57E-17 | 5.14E-05 |
| ILMN_1686623 | 1436 | CSF1R | -4.67 | -3.94 | -7.38 | -3.13 | 3.12E-16 | 6.55E-12 | 3.65E-24 | 1.54E-07 |
| ProbeID | EntrezID | SymbolID | fc_Borrelia_4h | fc_Candida_4h | fc_LPS_4h | fc_Mtb_4h | padj_Borrelia_4h | padj_Candida_4h | padj_LPS_4h | padj_Mtb_4h |
|---|---|---|---|---|---|---|---|---|---|---|
| ILMN_1772964 | 6355 | CCL8 | 1.98 | 27.48 | 1.62 | 1.76 | 4.52E-01 | 3.85E-23 | 1.00E+00 | 1.00E+00 |
| ILMN_1791759 | 3627 | CXCL10 | -1.11 | 14.24 | -1.25 | -1.18 | 1.00E+00 | 2.31E-19 | 1.00E+00 | 1.00E+00 |
| ILMN_1801307 | 8743 | TNFSF10 | 1.01 | 5.64 | 1.33 | -1.40 | 1.00E+00 | 1.01E-33 | 7.92E-01 | 7.82E-01 |
| ILMN_2148785 | 2633 | GBP1 | 1.36 | 5.06 | 1.72 | 1.20 | 1.00E+00 | 1.56E-13 | 1.76E-01 | 1.00E+00 |
| ILMN_1701114 | 2633 | GBP1 | 1.31 | 4.28 | 1.53 | 1.01 | 1.00E+00 | 6.70E-12 | 5.94E-01 | 1.00E+00 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| ILMN_1728236 | 55106 | SLFN12 | -1.06 | 1.20 | -1.04 | -1.05 | 1.00E+00 | 4.92E-02 | 1.00E+00 | 1.00E+00 |
| ILMN_1684634 | NA | RP3-365I19.1-001 | -1.06 | 1.20 | -1.02 | -1.02 | 1.00E+00 | 9.40E-03 | 1.00E+00 | 1.00E+00 |
| ILMN_2088990 | 80231 | CXorf21 | -1.02 | 1.19 | -1.00 | -1.03 | 1.00E+00 | 8.59E-03 | 1.00E+00 | 1.00E+00 |
| ILMN_1712035 | 11070 | TMEM115 | 1.01 | -1.26 | 1.01 | 1.05 | 1.00E+00 | 4.91E-03 | 1.00E+00 | 1.00E+00 |
| ILMN_1699253 | 7417 | VDAC2 | 1.01 | -1.27 | -1.02 | 1.01 | 1.00E+00 | 6.10E-04 | 1.00E+00 | 1.00E+00 |
Enrichment-Analyse
(Funktionelle Annotierung)
Schließlich sollen Gruppen von Genen dahingehend untersucht werden, inwieweit in ihnen Gene mit bestimmten Funktionalitäten überrepräsentiert sind. Damit kann bestehend auf dem umfangreichen und aktuellen Wissen für eine möglicherweise unbekannte Bedingung die Antwort auf funktionaler Ebene bestimmt werden. Dafür bietet sich die sogenannte Enrichment-Analyse an, siehe [9], welche in mindestens zwei Teilklassen bezüglich der "Testziele" unterschieden werden kann: (1) Verteilung und (2) Auftrittshäufigkeiten. Bei letzteren und im Folgenden werden verschiedene Testmethoden (χ²-Test, Hypergeometrischer Test, exakter Fisher-Test) auf Vierfeldertafeln (Konfusionsmatritzen) angewendet.
Die Grundlage des Enrichments bilden immer Datenbanken mit Annotierungen und somit der Bildung von Gruppen bzw. Kategorien. Die am häufigsten untersuchten Kategorien entstammen der "Gene Ontology"-Annotierung und der "KEGG-Pathways". Hier ist zu beachten, dass erstere die Form eines gerichteten azyklischen Graphen besitzt, bei dem Terme in untergeordneten Kategorien auch zu übergeordneten gehören (aber nicht anders herum). Daher ist es notwendig, diese Abhängigkeit mitzuberücksichtigen, wie es beispielsweise in [10] beschrieben wurde.
Die folgenden Abbildungen zeigen auf der Basis von Listen der differentiell veränderten Gene nach Candida-Infektion und der allgemein veränderten Gene einer Auswahl der wichtigsten Kategorien aus der Gene Ontology und von KEGG (für letztere wurde hier die Plattform FungiFun2 benutzt, siehe [11]):
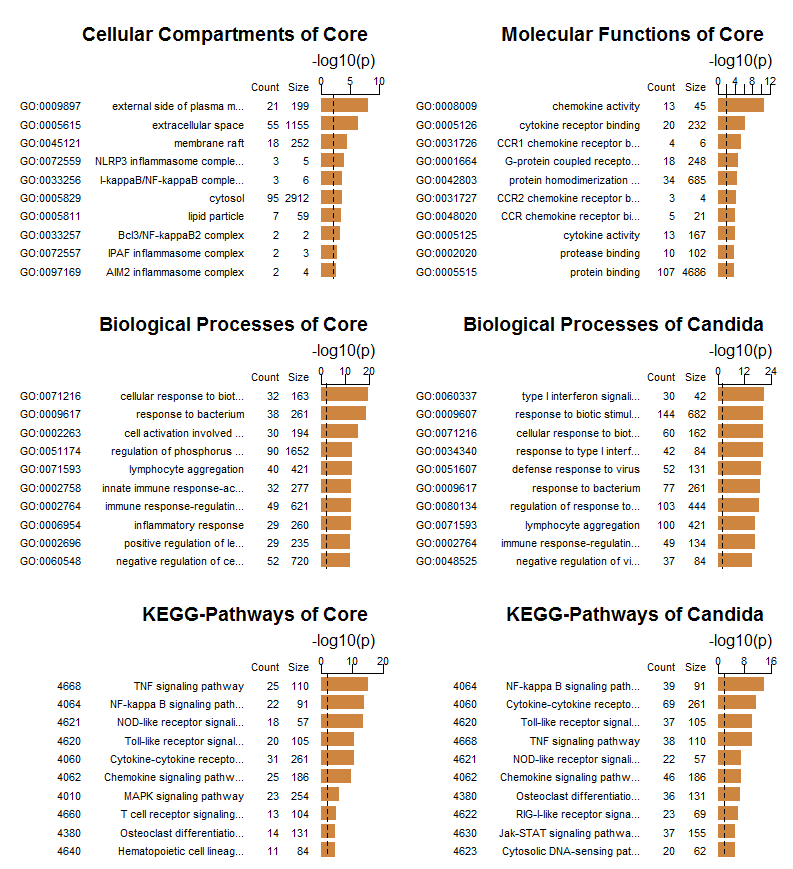
Die GO-Annotierungen bezüglich der zellulären Komponenten und der molekularen Funktionen bestätigen vor allem die Erwartungen. Als wichtigstes Ergebnis bei den biologischen Prozessen nach Candida-Infektion ist - wie auch in der ursprünglichen Publikation gezeigt - die Kategorie bezüglich Typ I Interferon hervorzuheben. Der Vergleich der KEGG-Pathways zeigt sehr schön die unterschiedlich aktiven Signalpfade für die allgemeine Antwort und nach Candida-Infektion. Weitere Interpretationen dieser Ergebnisse müssten dann gemeinsam mit Experten der Biologie und Medizin erfolgen.
(Auf Anfrage sind die vollständigen Ergebnisse bei BioControl verfügbar)
Referenzen
- [1] S. P. Smeekens, A. Ng, V. Kumar, M. D. Johnson, T. S. Plantinga, C. van Diemen, P. Arts, E. T. P. Verwiel, M. S. Gresnigt, K. Fransen, S. van Sommeren, M. Oosting, S.-C. Cheng, L. A. B. Joosten, A. Hoischen, B.-J. Kullberg, W. K. Scott, J. R. Perfect, J. W. M. van der Meer, C. Wijmenga, M. G. Netea and R. J. Xavier: Functional genomics identifies type I interferon pathway as central for host defense against Candida albicans. In: Nat Commun, 4:1342, 2013. doi: 10.1038/ncomms2343
- [2] J. Linde, S. Schulze, S. G. Henkel and R. Guthke: Data- and knowledge-based modeling of gene regulatory networks: an update. In: EXCLI Journal, 14:346–378, 2015.
doi: 10.17179/excli2015-168 - [3] M. Weber, S. G. Henkel, S. Vlaic, R. Guthke, E. J. van Zoelen and D. Driesch, Inference of dynamical gene-regulatory networks based on time-resolved multi-stimuli multi-experiment data applying NetGenerator V2.0. In: BMC Syst Biol, 7:1, 2013.
doi: 10.1186/1752-0509-7-1 - [4] B. M. Bolstad, R. A. Irizarry, M. Astrand and T. P. Speed: A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. In: Bioinformatics, 19(2):185–193, 2003. doi: 10.1093/bioinformatics/19.2.185
- [5] W. S. Togerson: Theory and methods of scaling. New York: J. Wiley, 1958. ISBN: 978-0471879459
- [6] D. J. McCarthy and G. K. Smyth: Testing significance relative to a fold-change threshold is a TREAT. In: Bioinformatics, 25(6):765–771, 2009. doi: 10.1093/bioinformatics/btp053
- [7] Y. Benjamini and Y. Hochberg: Controlling the False Discovery Rate: a practical and powerful approach to multiple testing. In: Journal of the Royal Statistical Society, Series B, 57(1):289–300, 1995.
- [8] M. E. Ritchie, B. Phipson, D. Wu, Y. Hu, C. W. Law, W. Shi and G. K. Smyth: limma powers differential expression analyses for RNA-sequencing and microarray studies. In: Nucleic Acids Research, 43(7):e47, 2015. doi: 10.1093/nar/gkv007
- [9] D. W. Huang, B. T. Sherman and R. A. Lempicki: Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. In: Nucleic Acids Research, 37(1):1–13, 2009. doi: 10.1093/nar/gkn923
- [10] S. Falcon and R. Gentleman: Using GOstats to test gene lists for GO term association. In: Bioinformatics, 23(2):257–258, 2007. doi: 10.1093/bioinformatics/btl567
- [11] S. Priebe, C. Kreisel, F. Horn, R. Guthke and J. Linde: FungiFun2: a comprehensive online resource for systematic analysis of gene lists from fungal species. In: Bioinformatics, 31(3):445–446, 2015. doi: 10.1093/bioinformatics/btu627